-
Email info@jnans.org
-
Address 848 N. Rainbow Blvd. #5486 Las Vegas, NV 89107, USA
1Department of Neurology, University of Tennessee Health Science Center College of Medicine, Memphis, TN, USA.
2Department of Neurology, Georgetown University Medical Center, Washington, DC, USA.
3National Institute of Neurological Disorders and Stroke, National Institutes of Health, Bethesda, MD USA.
4Department of Critical Care Medicine, Washington Hospital Center, Washington, DC USA.
5Department of Neurology and Neurosurgery, SemmesMurphey Neurologic and Spine Institute, Memphis, TN USA. China.
*Corresponding author: Nitin Goyal
University of Tennessee Health Science Center College of
Medicine 309 Hyman Admin Bldg, Memphis, TN 38163, USA.
Email ID: ngoyal@uthsc.edu
Tel: +1-901-758 7888
Received: Aug 14, 2025
Accepted: Oct 15, 2025
Published Online: Oct 22, 2025
Journal: Journal of Neurology and Neurological Sciences
Copyright: Goyal N et al. © All rights are reserved
Citation: Zambrano MDO, Kitani T, Chang JJ, Goyal N. Mechanisms of delayed cerebral ischemia after aneurysmal subarachnoid hemorrhage. J Neurol Neuro Sci. 2025; 1(2): 1010.
Background: The management of Aneurysmal Subarachnoid Hemorrhage (aSAH) in-volves monitoring with clinical exam, vital signs, and transcranial Doppler. “Delayed Cerebral Ischemia” (DCI) is characterized by clinical deterioration and evidence of stroke on imaging that occur beyond the first 72 hours after aSAH. Traditionally, treatment of DCI emphasizes optimization of cerebral perfusion pressure and cerebral blood flow.
Methods: We completed a review evaluating alternative mechanisms of secondary injury in aSAH that could present as therapeutic targets for mitigating DCI.
Results: Mechanisms for DCI are multifactorial. Aneurysm formation and subsequent aSAH involves a complex pathophysiology that includes endothelial damage and inflammation leading to DCI as a final common pathway. Multiple studies have focused on mechanisms of DCI that involve vasospasm, endothelial damage and microthrombosis, Cortical Spreading Depolarization (CSD), impaired cerebral autoregulation, and inflammation. Ultimately, these mechanisms cascade together to propagate DCI. Many studies in the past demonstrated failures of agents trying to address such singular causes of DCI.
Conclusion: We propose the use of biomarker monitoring as an additive to current monitoring techniques for patients at risk for DCI after aSAH. We also present a multimodal treatment approach for mitigating DCI in aSAH, that addresses these various mechanisms.
Keywords: Aneurysmal subarachnoid hemorrhage; Subarachnoid hemorrhage; Delayed cerebral ischemia; Mechanism; Intervention.
Aneurysmal Subarachnoid Hemorrhage (aSAH) is associated with significant morbidity and mortality (30-45%) [1]. It has a unique bimodal pathophysiology characterized by “Early Brain Injury” (EBI) that occurs within 72 hours of ictus and a subacute “vaso-spasm” phase, occurring around days 4-21 postictus, that can result in Delayed Cerebral Ischemia (DCI). DCI is a significant contributor to morbidity and mortality associated in aSAH [2]. DCI has a wide nomenclature—delayed ischemic deficit or symptomatic vasospasm—and has been loosely defined by the presence of clinical deterioration manifested by focal neurological deficits or presence of cerebral infarcts on brain imaging [3]. Traditionally, DCI was thought to be caused primarily by cerebral vasospasm. However, management of DCI by targeting cerebral vasospasm or optimizing cerebral perfusion pressure continues to demonstrate limited efficacy. Most recently, treatment of cerebral vasospasm with clazosentan [4] failed to demonstrate DCI prevention or improvement in functional outcome. This article aims to review potential proposed mechanisms and outline potential therapeutic targets to mitigate and treat DCI related to aSAH. This study was conducted according to the guidelines of the Declaration of Helsinki, and approved by the Institutional Review Board.
Aneurysm formation, Early Brain Injury, and Early Systemic Injuries The mechanism of intracranial aneurysm formation and subsequent disease progression is characterized by initial endothelial damage, followed by inflammation and microvascular changes (Figure 1). The formation of intracranial aneurysms begins with wall shear stress that leads to an inflammatory response in the endothelial cells of the vessel wall. This inflammatory response leads to further local damage (vascular smooth muscle cell apoptosis, extracellular matrix degradation, inflammatory cell infiltration) and ultimately leads to rupture, which is estimated to occur in 3-5% of the cases [1]. Notably, inflammatory cell infiltration is a hallmark of intracranial aneurysm formation, primarily spurred by macrophages [5].
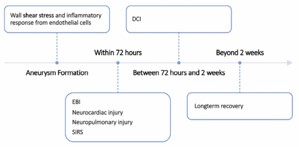
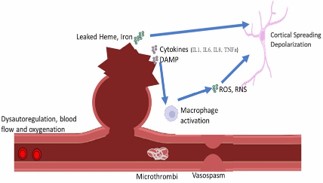
DCI represents a final common pathway for cerebral ischemia that results after aSAH. Although vasospasm has been the primary and likely most important target for mitigating DCI, recent studies have attempted to broaden the mechanisms associated with DCI and also to prognosticate which patients develop DCI [13-15]. The pathophysiological mechanisms that result in DCI can be broadly categorized as follows: vasospasm, endothelial damage and microthrombosis, impaired cerebral autoregulation, cortical spreading depolarization, and inflammation (Figure 2).
Following intracranial aneurysm rupture, EBI may occur within 72 hours and can be a strong predictor of clinical outcome in aSAH [6]. During the EBI phase, patients may develop cerebral edema, microthrombosis, inflammation, and spreading depolarization. These events in turn can lead to pathophysiologic responses including elevated Intracranial Pressure (ICP), microthrombus formation, and unregulated inflammatory response. In EBI, patients may also undergo cardiopulmonary injury triggered by adrenergic overload. This “neurogenic stunned myocardium” demonstrates electrocardiographic and echocardiographic abnormalities, as well as troponemia [7]. Pathophysiology centers around catecholamine release and myocardial microvascular dysfunction [7,8], which may be detected through highly sensitive strain imaging and may be associated with worse clinical outcomes [9]. Neurogenic pulmonary edema correlates with higher Intracranial Pressure (ICP) and can follow either neurocardiogenic injury or downstream adrenergic activation in the pulmonary system [10]. Both cardiac and pulmonary injury usually occur within 72 hours, concordant with the timing of peak catecholamine concentrations. Patients with aSAH also commonly demonstrate signs of diffuse inflammation, including fever, leukocytosis, tachycardia, and tachypnea (commonly grouped as Systemic Inflammatory Response Syndrome (SIRS) criteria). These signs are present in over half of patients at admission, reaching 60-80% within 4 days of aSAH onset [11]. The serum of EBI patients within 48 hours of admission were shown to have elevated Inflammatory and Macrophage-associated cytokines (IL6, MIP1B) [6]. Various theories have been posited to explain EBI’s association with the general inflammatory response and this can ultimately lead to the onset of cerebral vasospasm and DCI: Blood-Brain Barrier (BBB) disruption, brain tissue hypoxia, metabolic derangements, microthrombosis, free radical reaction, and vasoconstriction [12]. This spectrum of inflammatory responses and markers are all potential targets for future study as we work to understand the nature of DCI in aSAH.
Vasospasm
Vasospasm is widely recognized as the most common cause of Delayed Cerebral Ischemia (DCI) and is strongly associated with its occurrence [16]. Typically manifesting between 3 to 21 days after an initial aneurysmal rupture, up to 70% of patients with aSAH may exhibit cerebral vasoconstriction detectable through Digital Subtraction Angiography (DSA) [17]. The risk of vasospasm closely correlates with the volume of blood in the subarachnoid space and its proximity to the major vessels comprising the Circle of Willis, as well as the location of the aneurysm. Ruptured posterior circulation aneurysms are less likely to lead to DCI compared to aneurysms in the anterior circulation (Table 1) [14,18]. Monitoring for DCI in the intensive care unit predominantly focuses on the early detection of cerebral vasospasm, as ischemia resulting from large artery vasospasm remains the only potentially reversible cause of DCI with current therapeutic interventions [17]. In aSAH clinical trials, angiographic vasospasm is most commonly defined as a reduction in cerebral artery diameter by more than two-thirds of its original size [19]. However, the degree of angiographic narrowing does not always strongly correlate with clinical symptoms or outcomes. Interestingly, while up to 70% of patients with SAH may demonstrate vasoconstriction or vasospasm on angiography, only about 30% of them develop clinical symptoms attributable to ischemia caused by vasospasm [20]. This indicates that angiographic vasospasm may be clinically asymptomatic in many cases, emphasizing the importance of vigilant neurological monitoring and assessment.
Treatment is generally indicated when patients present with neurological symptoms suggestive of ischemia. To optimize care, prediction methods are employed to stratify patients based on their risk of developing DCI. Tools such as the Modified Fisher Scale and VASOGRADE are commonly used to identify high-risk individuals who may benefit from aggressive monitoring and timely therapeutic interventions [18]. These approaches aim to balance the need for close observation with the judicious use of resources, ultimately improving outcomes for patients at risk of DCI. Imaging modalities such as Computed Tomography (CT) angiography and DSA are commonly utilized for the detection of vasospasm in patients with aSAH. However, their frequent use is discouraged due to the cumulative risks associated with repeated exposure to radiation and contrast agents. Moreover, these imaging techniques are limited in their ability to provide dynamic or real time monitoring of vascular changes, which is often essential for timely diagnosis and intervention in critically ill patients [21]. As an alternative, Transcranial Doppler ultrasound (TCD) has become a standard first-line ancillary tool for monitoring vasospasm, particularly in the large arteries adjoining the Circle of Willis. TCD is recognized for its high sensitivity in detecting changes in the Middle Cerebral Artery (MCA) and the basilar artery [21,22]. Research has demonstrated that a TCD-recorded MCA peak flow velocity of 200 cm/s or higher, when combined with epileptiform abnormalities detected on Electroencephalography (EEG), offers a stronger predictive value for the onset of Delayed Cerebral Ischemia (DCI) compared to either modality used independently [23]. To address the challenge of distinguishing vasospasm from hyperemia, the Lindegaard ratio is commonly employed as a normalizing metric. This ratio is calculated by dividing the mean flow velocity of the MCA by the mean flow velocity of the terminal internal carotid artery. A Lindegaard ratio below 3 typically suggests hyperemia, while a ratio greater than 3 indicates vasospasm. Further stratification of this ratio allows clinicians to classify the severity of vasospasm: A ratio greater than 3 indicates mild vasospasm, greater than 4 suggests moderate vasospasm, and greater than 5 reflects severe vasospasm. These diagnostic tools and criteria enhance the ability to identify and manage vasospasm [24].
The intra-arterial administration of vasodilators, often combined with or without angioplasty, is a widely used treatment for vasospasm linked to DCI. Clinical trials involving nimodipine [25,26] and intra-arterial verapamil [27] have demonstrated some benefit in managing vasospasm following aSAH. Additionally, recent trials, such as the CONSCIOUS-1 trial evaluating clazosentan—a more potent vasospasm-combative medication—have shown a significant reduction in vasospasm events. However, this reduction did not translate into a notable improvement in DCI outcomes [28,29]. Interestingly, subgroup analyses from the study revealed that patients with poor-grade aSAH experienced better outcomes with clazosentan compared to placebo. These findings reaffirm the role of vasospasm in contributing to DCI after aSAH but highlight that vasospasm alone does not entirely explain the complex pathophysiology of DCI. Another agent commonly utilized is milrinone [30], which acts as a vasodilator but is associated with a potential risk of hypotension. Beyond pharmacological interventions, balloon angioplasty has been explored as a treatment option for refractory vasospasm. While limited studies have shown that balloon angioplasty can improve vasospasm severity without causing complications, approximately 20% of the brain territories supplied by the treated vessel still develop infarcts [31]. These findings underscore the need for further research to optimize both pharmacologic and interventional approaches to managing vasospasm and its related complications.
| (Lee et al, 2019) | (Göttsche et al, 2022) | ||||
|---|---|---|---|---|---|
| Aneurysm Location | DCI (OR) | p-value | DCI (OR) | p-value | Comments on Outcome Impact |
| Anterior Communicating Artery (ACom) | 2.5-3.2 | <0.01 | 2.8-3.5 | <0.01 | Highest DCI risk; commonly affects ACA territory, leading to cognitive and executive dysfunction. |
| Middle Cerebral Artery (MCA) | 2.0-2.8 | <0.05 | 2.3-3.0 | <0.05 | High risk of vasospasm and ischemia, often causing motor deficits and aphasia. |
| Internal Carotid Artery (ICA) Posterior Communicating Artery (PCom) | 1.8-2.5 | 0.05 –0.10 | 1.9-2.7 | <0.05 | Moderate risk; affects MCA and anterior choroidal artery,leading to visual field deficits and hemiparesis. |
| Basilar Artery (BA) / Vertebrobasilar System | 0.9-1.5 | >0.10 (NS) | 1.1-1.6 | 0.05 -0.10 | Lower risk but severe consequences, (NS) 1.6 0.10 including brainstem infarcts and high mortality. |
| Posterior Cerebral Artery (PCA) | 0.7-1.3 | >0.10 (NS) | 0.8-1.4 | – >0.10 (NS) | Lowest DCI risk; may result in visual impairments if affected. |
Endothelial damages and microthrombosis
Vascular dysfunction centers around platelet aggregation and endothelial damage. Platelet aggregation and vasospasm in intraparenchymal arterioles cause micro-clot formation, a process known as microthrombosis [32]. Endothelial damage leads to platelet aggregation and leukocyte adhesion [33]. Cellular aggregation combined with micro-thrombosis can then lead to a worsening clot and lead to a higher ischemic burden in the cerebral vasculature.
Platelet adhesion and thrombosis-induced inflammatory responses are maintained by the activity of the endothelial protease ADAMTS13. Development of DCI correlated with decreased ADAMTS13 activity and increased platelet activation markers such as von Willebrand factor and P-selectin. Preclinical data has demonstrated the importance of P-selectin expression in the microvascular endothelium after aSAH [32], and a reduction in the levels of P-selectin led to decreased platelet-endothelial and leukoendothelial interactions [33]. Additionally, D-dimer level was increased in aSAH and higher levels correlated with high risk of DCI in Fisher grade 4 aSAH [34,35].
Cerebrospinal fluid markers may also be associated with endotheliopathy in aSAH [36]. In a small study evaluating cerebrospinal fluid in aSAH, DCI correlated with elevated cell adhesion molecule-1 and increased cerebrospinal fluid matrix metalloproteinase.
Small trials have demonstrated that anticoagulation may reduce DCI following aSAH. Meta analyses, as well as retrospective studies, showed antiplatelet agents (aspirin, clopidogrel) may be useful in decreasing the incidence of DCI but noted the increased risk of hemorrhagic complications [37,38].
Impaired cerebral autoregulation
Cerebral Autoregulation (CA) maintains an adequate and constant Cerebral Blood Flow (CBF), regardless of arterial blood pressure fluctuations. This regulation is achieved through a complex interplay of metabolic, endothelial, and neurogenic responses, which collectively work to maintain homeostasis and prevent ischemic or hyperemic damage to the brain [39]. However, in patients with aSAH, this autoregulatory capacity is often compromised [40]. The impairment of CA in aSAH has been linked to the development of DCI, although the extent and significance of its contribution remains debatable in the literature. Studies have reported mixed findings regarding the precise role of impaired CA in the pathophysiology of DCI, highlighting the complexity of the condition. To evaluate CA, several indices are commonly utilized in clinical and research settings. These include Pressure Reactivity (PRx)—measuring the correlation between intracranial pressure and mean arterial pressure, Oxygen Reactivity (ORx)—measuring the relationship between brain oxygen levels and mean arterial pressure, and Flow Reactivity (FRx)—measuring the dynamic responses of blood flow to changes in perfusion pressure. These indices provide valuable insights into the functionality of CA and help clinicians better understand its role in the progression and management of DCI in aSAH patients.
The findings from studies evaluating traditional indices of CA have been inconsistent. Two investigations demonstrated a strong correlation between the ORx and clinical outcomes in patients with aSAH at six months [40] and with the occurrence of DCI [41]. Conversely, a separate study failed to establish a similar association [42]. Notably, a correlation between ORx and FRx was observed across these studies, suggesting a link between these autoregulation metrics. The collective evidence implies that DCI may develop when cerebral autoregulation fails to compensate for the vasoconstriction induced by vasospasm [43]. These findings underscore the need for further research to clarify the role of autoregulatory dysfunction in the pathophysiology of DCI and to optimize clinical monitoring and management strategies.
To address DCI caused by CA, the “Triple H” therapy—comprising hypertension, hemodilution, and hypervolemia—was historically employed. However, these interventions were often associated with significant complications and yielded only minimal improvement in clinical outcomes [44,45]. Consequently, current therapeutic approaches now prioritize blood pressure support rather than implementing aggressive hyper-volemia or hemodilution [46]. Emerging evidence from small clinical trials suggests that systemic erythropoietin and pravastatin may enhance CA, potentially offering alternative therapeutic strategies [47]. These findings support the hypothesis that low ORx and FRx could be improved through interventions that increase hemoglobin levels, leading to induced hypertension. This approach may present a more favorable complication profile compared to therapies involving exogenous sources. Further studies are warranted to confirm these observations and optimize treatment protocols.
Cortical spreading depolarization
A metabolic mismatch can occur after aSAH, resulting in Reactive Oxygen Species (ROS) formation, increased Reactive Nitrogen Species (RNS), along with accumulation of other metabolic waste. This metabolic waste combined with hemolyzed blood triggers massive neuronal death, and further hypoperfusion of the brain tissue leading to a “snowball effect” of neuronal damage [48]. Cortical Spreading Depression (CSD), a sign of brain tissue impairment, has been theorized to precede and lead to DCI, even in the absence of angiographic vasospasm [49].
Experimentally, onset of CSD is attributed to disruption of the neuronal environment leading to glutamate-induced toxicity, resulting in failure for ion homeostasis by ion channels, and subsequent fluid shifting and neuronal swelling [50]. The neuronal environment disruption is attributed to two main pathways: the interaction between iron and heme with lipid coating the neurons, and inflammatory responses by immune cells resulting in release of ROS and RNS [48]. This data highlights an important concept observed in DCI: mechanisms that cause DCI also contribute to other mechanisms that lead to DCI. As a result, it can be quite challenging to separate the myriad of pathophysiological mechanisms involved in vasospasm, CA, CSD, and inflammation.
Given that the CSD mechanism involves excitotoxicity and ROS/ RNS formation, basic science experiments have offered potential therapies for DCI prevention. A canine aSAH DCI model used edavarone, a free radical scavenger, to demonstrate decreased basilar artery narrowing [51]. Edavarone was also utilized in a small human trial of 91 patients with aSAH. This study showed edavarone resulted in a significant reduction in DCI from vasospasm and mitigated poor outcomes caused by vasospasm [52].
Inflammation and blood-brain barrier breakdown
Inflammation plays a key role in aSAH. Another proposed mechanism of inflammation in DCI is the activation of Toll-Like Receptor (TLR) via metabolites from blood and hemoglobin, leading to activation of macrophages and release of inflammatory cytokines [11]. One such reactant is fibrinogen, which at baseline is found in low levels in the CSF but is increased in the setting of blood-brain barrier disruptions as seen in aSAH [53].
Additionally, inflammation is further exacerbated via Damage-Associated Molecular Pattern (DAMP) released upon injured cells, resulting in activation of macrophages via TLR-pathway and leading to additional injury [54]. An example of DAMP association with DCI is peroxiredoxin-2, which is a component of erythrocytes released during aSAH and found to be at high levels in the CSF after aSAH. Peroxiredoxin-2 interacts with TLR4 and activates macrophages that serve a substantial a role in DCI [55].
Systemic inflammation following aSAH, as demonstrated by various factors in-cluding C-reactive protein, erythrocyte sedimentation rate, leukocyte count, and lactate concentration, also seem to correlate with DCI and poorer outcomes [56-58]. Inflammatory cytokines in CSF, such as IL1, IL6, IL8, TNFa are associated with worse prognosis in aSAH patients [59]. Specifically, IL6 in CSF was measured at a higher level than in serum, and higher CSF IL6 levels were associated with DCI [60]. One marker of systemic inflam-mation that may help prognosticate DCI occurrence and clinical outcomes after aSAH may be neutrophil-to-lymphocyte ratios [61-63]. Some therapeutic agents that have been evaluated in this context include steroids [64, 65] and nonsteroidal anti-inflammatory drugs [66]. Currently, however, there are no large-scale trials that have examined the utility of these agents in the management of aSAH.
| Vasospasm | |||
|---|---|---|---|
| Author. year | Study type | Intervention | Results |
| Pala A et al. [25] | Prospective Human Study | Intracarotid Arterial Nimodipine | Resolution of DCI symptoms in study subjects [18]. |
| Pickard JD et al. [26] | Double blind, placebo controlled, Randomised trial | Oral Nimodipine 60 mg daily | Significant decrease in incidence of cerebral infarction and poor outcomes [19]. |
| Sehy JV et al. [27] | Retrospective observational case series | Intra-arterial verapamil | Improved angiographic vasospasm [20]. |
| Macdonald RL et al. [28] | Randomized Controlled Trial | IV clazosentan | Clazosentan decreased moderate and severe vasospasm in a dose-dependent manner, and reduction in vasospasm- related morbidity/mortality [21]. |
| Macdonald RL et al. [29] | Double blind, placebo controlled, randomised trial | IV clazosentan | No significant effect on mortality and vasospasm-related morbidity or functional outcome [22]. |
| Castle-Kirszbaum M et al. [30] | Pooled systemic Review | IV or intra-arterial milrinone | IV or intraarterial-intravenous milrinone led to clinical resolution of DCI in above 80% of cases, with cerebral infarctions attributable to DCI occurred in 19%. However, hypotension occurred in 23% and hypokalemia occurred in 11% as common side effects [23]. |
| Patel AS et al. [31] | Retrospective study | Balloon angioplasty | Balloon angioplasty improved the severity of vasospasm in 97% of vessels without complications; however, approximately 20% of territories supplied by vessel undergoing angioplasty experienced infarcts [24]. |
| Endothelial damage and microthrombosis | |||
| Dorhout Mees SM et al. [37] | Systematic Review | Oral Aspirin | Trend towards better outcome that was not statistically significant, and with increased hemorrhagic complications [30]. |
| Nagahama Y et al. [38] | Retrospective observational case series | Dual antiplatelet therapy | Dual Antiplatelet therapy was associated with lower risk of clinical vasospasm and DCI, without increased risk of hemorrhagic complications [31]. |
| Impaired cerebral autoregulation | |||
| Dankbaar JW et al. [44] | Systematic Review | Hemodilution, Hypervolemia, Hypertension | No evidence supported the use of triple-H therapy on cerebrovascular blood flow in aSAH patients [37]. |
| Le Roux PD et al. [45] | Literature Review | Red Blood Cell (RBC) Transfusion | RBC transfusion was associated with medical complications, infection, vasospasm, and poor outcome after aSAH [38]. |
| Tseng MY et al. [47] | Randomized, Double-blind, Place-bo- controlled Trial | Systemic Erythropoieitin (EPO) Therapy | EPO therapy delayed cerebral EPO therapy delayed cerebral |
| Cortical spreading depolarization | |||
| Nakagomi T et al. [51] | Canine SAH model | Edavarone, bolus or continuous infusion to IV | IV or continuous infusion of Edavarone was associated with attenuated narrowing of basilar artery following SAH [44]. |
| Munakata A et al. [52] | Prospective Human Study | IV Edavarone | Trend toward a lesser incidence of DINDs and a lesser incidence of poor outcome caused by cerebral vasospasm in edaravone-treated patients [45]. |
| Inflammation and BBB breakdown | |||
| Chyatte D et al. [64] | Matched case study | IV Methylprednisolone | The incidence and severity of delayed cerebral ischemia were reduced in treated patients when compared to control patients. None of the treated patients developed a serious side effect that could be attributed to steroid treatment [57]. |
| Hashi K et al. [65] | Double blind, placebo controlled, randomised trial | Double blind, placebo controlled, randomised trial | In patients with Fisher grade I- III on admission, the steroid- treated group had favorable effects on neurological outcomes (mental, speech, motor function) than in placebo group [58]. |
| Solar P et al. [66] | Systematic Review | NSAID use in aSAH in clinical context or mouse experimental model context | NSAID theoretically may reduce aSAH induced vasospasm. but only one RCT demonstrated tendency towards better outcome of NSAID use in aSAH [59]. |
| Multiple mechanism | |||
| Porchet F et al. [68] | Case series | IV Nimodipine | No significant difference of incidence of DCI. and risk for hypotension exists [61]. |
| Choi HA et al. [69] | Observational study | Oral Nimodipine | Oral nimodipine was associated with decrease in MAP. CPP. decreased Pox and CBF [62]. |
| Can A et al. [70] | Case-Control study | Oral Aspirin | Aspirin therapy was associated with a significantly decreased risk of aSAH. but when once ruptured Aspirin use was associated with increased risk of rerupture [63]. |
| Kole MJ et al. [71] | Cohort comparison study | Low dose IV heparin vs heparin for deep vein thrombosis prophylaxis in aSAH whose aneurysm once secured by clipping or coiling | Low dose IV heparin infusion improved the outcome of patients [64]. |
| Pascale CL et al. [74] | In-vitro and mouse model | Dimethyl fumarate added to medium or Oral dimethyl fumarate | Dimethyl fumarate protected the vascular smooth muscle cells in- vitro from TNF-a induced inflammation. and inhibited the proliferative effect of TNF-a resulting in protection from aponecrosis. Dimethyl Fumarate demonstrated neuroprotective effect in mice with inhibition of oxidative stress. inflammation. and fibrosis in the cerebrovasculature [67]. |
Medications affecting multiple mechanisms
Some medications have multiple mechanisms of action that may be beneficial in the management of DCI. Nimodipine, a dihydropydirine calcium antagonist, is the current mainstay of management. Nimodipine has been shown to increase endogenous fibrinolysis [67]. However, nimodipine can increase the risk of secondary ischemia via arterial hypotension [68], and there is no convincing evidence that it would decrease cerebral vasospasm or DCI [69]. Aspirin a well-known antiplatelet agent may also act as an an-ti-inflammatory medication through its effect on the COX2 pathway [11]. However, some case-control studies have shown increased risk of aneurysmal re-rupture [70]. A retrospective study evaluating low dose heparin infusion in aSAH after securing the aneurysm with clipping or coiling demonstrated improved DCI and minimal iatrogenic bleeding [71]. Heparin has known impact on multiple pathways involved in aSAH such as inflammation and neutralization of reactive oxygen species, making it an attractive an-ti inflammatory therapy for minimizing DCI.
Another potential medication that addresses multiple mechanisms for DCI is Di-Methyl Fumarate (DMF), an immunosuppressant used in relapsing multiple sclerosis. In the context of multiple sclerosis, DMF is shown to reduce CD8+ T-cells and memory immune cells via DMF-induced apoptosis [72]. However, it is also a potent neutralizer of ROS via activation of the Nuclear Factor erythroid 2-related Factor 2 (Nrf2) pathway [73]. DMF was shown in-vitro, as well as in mouse models of aSAH, to decrease TNFa-induced inflammation along with aneurysm formation and rupture [74], making it a reasonable and attractive candidate for use in aSAH. (Table 2) provides a summary of the various potential therapeutic medications in aSAH.
Longitudinal studies have shown that the majority of aSAH patients who survive past the first 2 weeks after aSAH onset will survive to the five-year mark [75] and be functionally independent [76]. This potential long-term functional recovery makes mitigating secondary injury extremely important. We have discussed the multiple and varied pathophysiologies that contribute to DCI following aSAH. Many of the mechanisms of aSAH play a role in exacerbating other pathophysiological mechanisms and has a cascading deleterious effect.
Many of the pharmacological approaches of DCI address only solitary mechanisms in DCI. We feel that potential therapeutics that offer a more pleiotropic treatment regimen have not been adequately explored. Some of the medications discussed in this review have off-target mechanisms that may have far-reaching beneficial effects, potentially ameliorating multiple aspects of DCI.
One important initial step in evaluating these multifactorial mechanisms may be to gain a better understanding of prognostication after aSAH. This would provide insight into which of the proposed mechanisms have significant associations with DCI, and, ultimately, aid in developing more appropriate treatment regimens. We propose that this can be achieved by measuring commonly available serum markers that encompass the varied pathophysiologies of aSAH, including D-dimer, CRP, ESR, NLR, fibrinogen, fi-brin, and prothrombin. We also propose monitoring CSF markers such as IL6, TNFa and Tenascin-C, as well as CSF protein levels in order to develop a better understanding of which patients will go on to develop DCI [77]. We can then use these findings to better evaluate the efficacy of future multifactorial therapeutics targeting the mechanisms described in this review.
